Болезнь Помпе: вместе мы сильные
- 15 апреля 2018
- administrator

15 апреля во всем мире пациентские организации и пациенты с болезнью Помпе проводят образовательные и информационные кампании для повышения осведомлённости широкой общественности об этом редком наследственном заболевании с аутосомно-рецессивным механизмом
15 апреля во всем мире пациентские организации и пациенты с болезнью Помпе проводят образовательные и информационные кампании для повышения осведомлённости широкой общественности об этом редком наследственном заболевании с аутосомно-рецессивным механи
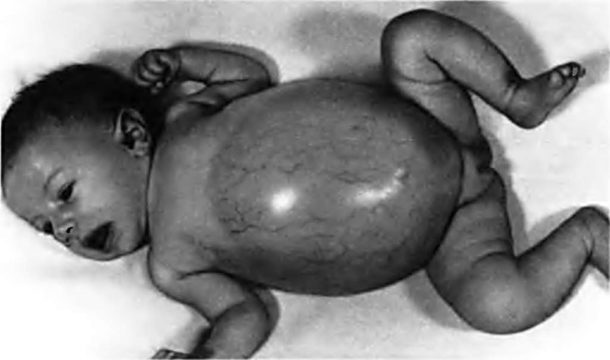
Ребенок с болезнью Помпе
Точная распространенность болезни Помпе неизвестна. По данным разных авторов, частота болезни, в зависимости от страны и этнической принадлежности, варьирует в диапазоне от 1:40.000 до 1:300.000.[1]При данном диагнозе в результате нехватки определенного фермента в клетках мышц и сердца больного накапливается особое вещество гликоген[2].
В семьях, где родители являются носителями болезни Помпе, риск рождения больного ребенка составляет 25% на каждую беременность.[3] В случае, если одному ребенку поставили этот диагноз, это не значит, что все дети будут больны, но нужно обязательно обследовать всех братьев и сестер.
Характерный признак заболевания – прогрессирующая мышечная слабость, в том числе скелетной и дыхательной мускулатуры[4]. Поражение сердечной мышцы проявляется в основном в начале заболевания в очень раннем возрасте. Такая форма заболевания называется младенческой. Она быстро прогрессирует и приводит к гибели на первом году жизни.5 В случаях юношеской или взрослой форм прогрессирование болезни включает в себя нарастающую слабость мышц туловища и ног, нарушение походки, одышку, дыхательную недостаточность, затруднение при жевании и глотании, вследствие быстрой утомляемости жевательных мышц. Без терапии, с течением времени, симптомы болезни нарастают. Многие пациенты перестают самостоятельно ходить и постепенно становятся зависимыми от аппарата искусственной вентиляции легких, поскольку происходит поражение мышц, ответственных за дыхание. [5], [6]
В отличии от многих других редких заболеваний, для пациентов с болезнью Помпе существует ферментозаместительная терапия, эффект от которой во многом зависит от того, когда пациент начал получать лечение. При своевременном и регулярном получении недостающего фермента, можно остановить или замедлить прогрессирование заболевания, что позволяет людям с этой болезнью вести активную социальную жизнь.
Ключевую роль в лечении болезни Помпе занимает ранняя диагностика.
«При диагностике редких заболеваний в целом и болезни Помпе в частности большая ответственность лежит на врачах первичного звена. Очень важно чтобы врач вовремя заподозрил «редкий диагноз» и направил пациента на обследование в случае необходимости. Но и сам пациент тоже должен быть довольно настойчивым и в тоже время иметь терпение, чтобы найти причину болезни и возможно провести обследование всей семьи. Нужно понимать, что нет «орфанного» врача, есть врач, который хочет разобраться. В среднем около 7 врачей должен обойти пациент с редким заболеванием, прежде чем ему поставят диагноз. И это не только в России, это во всем мире!», - подчеркнула Е. Ю. Захарова, заведующая лабораторией наследственных болезней обмена веществ Медико-генетического научного центра РАН, доктором медицинских наук.
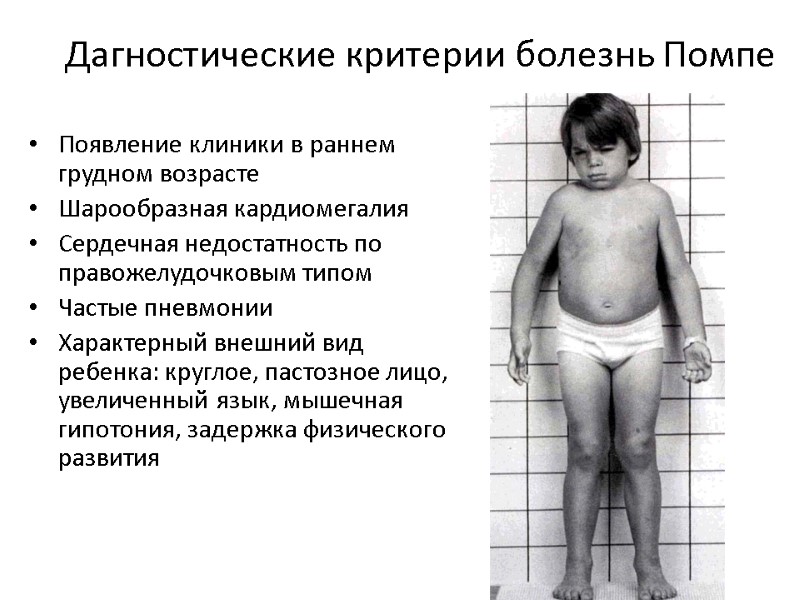
«Первые изменения в организме дочери мы заметили во время диспансеризации по ДМС. Процесс постановки диагноза был долгим, - отмечает Е., мама пациентки с болезнью Помпе, - Сначала ставили недифференцированный гепатит, но лечение не помогало. Потом нашли доктора, которая и помогла поставить диагноз – болезнь Помпе. В общей сложности мы пытались установить диагноз полтора года. После начался долгий период принятия диагноза, изучения информации и адаптации к жизни с болезнью.»
В России заболевание считается редким (орфанным), если его распространенность встречается не более 10 случаев на 100 000 населения.[7] Чаще всего они являются генетическими и проявляются в детстве7, но также дебют заболевания может произойти во взрослом возрасте. При адекватном лечении и систематическом применении лекарственных препаратов люди с орфанными заболеваниями могут вести активную жизнь и быть полноценными членами общества.[8]
[1] http://www.pediatr-russia.
[2] Hirschhorn, Rochelle and Arnold J. J. Reuser. Glycogen Storage Disease Type II: Acid Alphaglucosidase (Acid Maltase) Deficiency. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th Edition. New York: McGraw-Hill, 2001. 3389-3420.
[3] http://pompe.rare-diseases
[4] Kishnani PS, Steiner RD, Bali D, et al. Pompe disease diagnosis and management guideline. Genet Med 2006; 8:267-88
[5] Van den Hout HM, Hop W, van Diggelen OP, et al. The natural course of infantile Pompe's disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003; 112:332-40.
[6] Van der Beek Naet al. Clinical features and predictors for disease natural progression in adults with Pompe disease: a nationwide prospective observational study. Orphanet J Rare Dis. 2012 Nov 12;7:88
[7] http://spiporz.ru/redkie-z
[8] http://philanthropy.ru/int